ОРИГИНАЛЬНОЕ ИССЛЕДОВАНИЕ
Ретинальные аномалии у трансгенных мышей, суперэкспрессирующих аберрантный человеческий ген FUS[1-359]
1 Белгородский государственный национальный исследовательский университет, Белгород, Россия
2 Институт физиологически активных веществ, Москва, Россия
3 Курский государственный медицинский университет, Курск, Россия
4 Первый Московский государственный медицинский университет имени И. М. Сеченова, Москва, Россия
Повреждение сетчатки является неклассическим симптомом ряда нейродегенеративных заболеваний, включая боковой амиотрофический склероз (БАС). Целью работы было оценить морфофункциональное состояние сетчатки в мышиной модели БАС, связанной с суперэкспрессией аберрантного белка FUS[1-359]. Исследование проводили на 12 трансгенных и 13 диких мышах 2,5–3-месячного возраста. Выявлено, что трансгенные мыши демонстрируют выраженную, но не достигающую статистической значимости тенденцию к развитию офтальмоскопических аномалий сетчатки. Кроме того, молекулярно-биологический анализ подтвердил увеличение экспрессии провоспалительных генов Vegfa, Il1b, Il6, Icam1, Tnfa. При этом, несмотря на обнаруженные структурные и функциональные аномалии, вестерн-блот анализ и количественная ПЦР не выявили экспрессию белкового и мРНК продукта трансгена FUS в сетчатке мутантных мышей.
Ключевые слова: воспаление, белок FUS, трансгенные мыши, БАС, ретинопатия
Финансирование: исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта №19-315-90114.
Вклад авторов: В. О. Солдатов — разработка идеи и дизайна эксперимента, проведение офтальмоскопии, написание статьи, подбор праймеров для оценки экспрессии генов; М. С. Кухарский — разработка идеи и дизайна эксперимента, написание статьи, проведение иммуноблотинга; М. О. Солдатова — выделение РНК, проведение ПЦР в реальном времени; О. А. Пученкова — забор материала для молекулярно-биологических исследований, выделение РНК, проведение ПЦР в реальном времени; Ю. А. Никитина — подготовка популяции животных, генотипирование, проведение иммуноблотинга; Е. А. Лысикова — подготовка популяции животных, генотипирование, написание статьи; Карташкина Н. Л. — описание офтальмоскопической картины, написание статьи; А. В. Дейкин — консультация по основной идее и дизайну исследования; М. В. Покровский — консультация по основной идее и дизайну исследования.
Соблюдение этических стандартов: исследование одобрено локальным этическим комитетом Белгородского гоударственного национального исследовательского университета (протокол № 5/19–25 от 25 сентября 2019 г.), проведено с соблюдением требований Международных рекомендаций Европейской конвенции по защите позвоночных животных, используемых при экспериментальных исследованиях (1997).
Для корреспонденции: Владислав Олегович Солдатов
ул. Победы, д. 85, 308015, г. Белгород; moc.liamg@sregnifkniz
Сетчатка является наиболее доступным для изучения отделом нервной системы и одной из самых уязвимых сенсорных тканей. Подобные свойства актуализировали изучение офтальмологических коррелятов неврологических заболеваний, открывая возможности для улучшения диагностики и изучения нейродегенеративных процессов. За счет эмбриональной общности и схожести протеомного состава сетчатка может выступать платформой для развития тех же патологических каскадов, что и центральная нервная система [1]. В частности, повреждение сетчатки было обнаружено при боковом амиотрофическом склерозе (БАС) [2, 3], заболеваниии, для которого характерна прогрессирующая гибель мотонейронов вследствие накопления нерастворимых белковых агрегатов [4].
Белковые включения при БАС имеют сложный состав и могут состоять из различных белков, среди которых чаще всего обнаруживают РНК-связывающие белки или антиоксидантный фермент супероксиддисмутазу 1, а также другие компоненты: нейрофиламенты, убиквитин [5]. В 5% семейных случаев БАС ассоциирован с накоплением агрегатов, основным компонентом которых является белок FUS. Одна из причин развития FUS-протеинопатии — мутации в домене сигнала ядерной локализации (NLS) и выход белка из ядра в цитоплазму, где он приобретает способность к формированию нерастворимых агрегатов [6].
Цель данного исследования — оценить взаимосвязь между нейрональной экспрессией патологической формы белка FUS и активацией некоторых патологических путей в сетчатке.
МАТЕРИАЛЫ И МЕТОДЫ
Животные
В качестве модельного объекта FUS-протеинопатии были использованы мыши, несущие трансген аберрантного человеческого гена FUS c искусственно укороченным NLS (кодирует укороченный FUS[1-359]) [7]. Данная модель характеризуется манифестирующей в возрасте 3–4 месяцев клинической картиной БАС, которая сопровождается развитием морфологических и молекулярных признаков нейродегенерации, включая гибель нейронов и нейровоспаление [8].
Исследование проводили на 25 мышах (обоего пола) линии CD-1, 12 из которых были гемизиготами FUS, а 13 служили контролем дикого типа. Мышей содержали в условиях постоянного доступа к воде и корму. Световой цикл — 12 ч/12 ч, интенсивность освещения — 40–50 лк, температура воздуха — 23 ± 1 °С, влажность — 42 ± 5%. В возрасте 80–90 дней мышей седатировали (золазепам + тилетамин + ксилазин) для проведения офтальмоскопии. Перед седацией проводили клинический осмотр для исключения животных с признаками воспалительных изменений наружной камеры глаза.
Офтальмоскопическое исследование проводили после аппликации 1%-го атропина сульфата. Для объективизации анализа офтальмолог оценивал картину глазного дна по балльной шкале от 0 до 5, где 0 баллов — отсутствие нарушений, а 5 баллов — грубые аномалии. Данные офтальмоскопического анализа представлены в виде M ± SD, проверку статистической значимости межгрупповых различий проводили по критерию Краскелла–Уоллиса. После офтальмоскопического исследования животных эвтаназировали передозировкой наркоза и отбирали биоматериал для молекулярного анализа: у 6 животных из каждой группы извлекали образцы сетчатки для изучения генной экспрессии, а у 4 собирали образцы сетчаток и люмбального отдела спинного мозга для проведения вестерн-блотинга.
Количественная ПЦР
Ткани контрлатеральных сетчаток от каждого животного пуллировали и инкубировали 15 мин в растворе ExtractRNA («Евроген»; Россия). После лизирования образца в реагенте его подвергали хлороформной экстракции, а образовавшийся осадок РНК промывали последовательно изопропиловым спиртом и 70%-ным этиловым спиртом. Полученный осадок разбавляли в 20 мкл воды и с использованием спектрофотометра IMPLENNanoPhotometer® (Implen; Германия) измеряли концентрацию полученной РНК (~200 нг/мкл). Обратную транскрипцию проводили с использованием набора MMLVRTSK021 в соответствии с протоколом фирмыпроизводителя (Евроген; Россия).
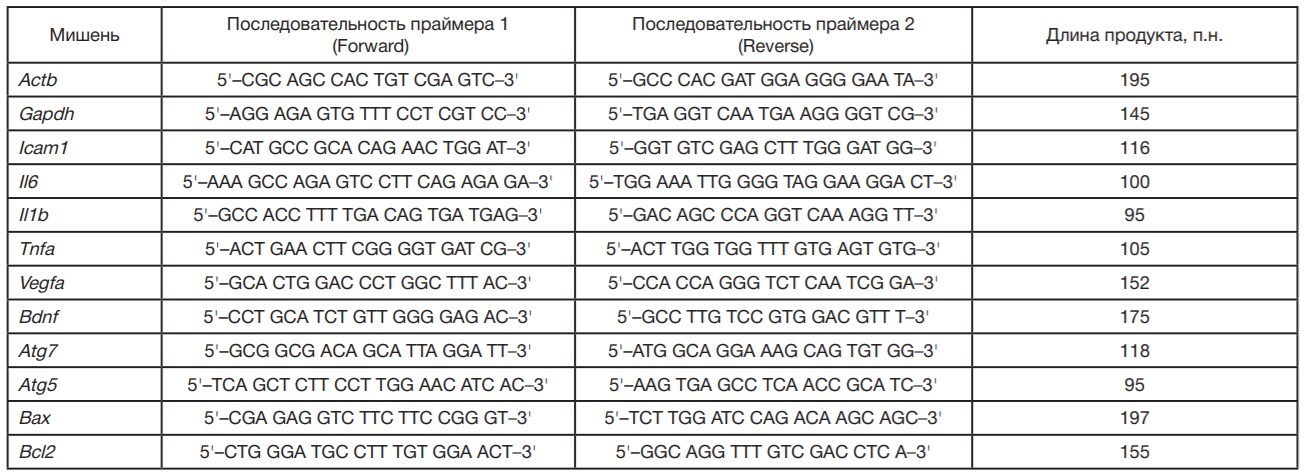
Праймеры для количественной ПЦР подбирали с использованием ресурса Primer-BLAST (NCBI) с соблюдением ряда условий: 1) температура плавления 59–61 °C; 2) один из праймеров в паре должен отжигаться на область межэкзонного соединения; 3) прямой и обратный праймеры не должны образовывать ауто- и кроссдимеров в одной смеси; 4) размер ПЦР-продукта должен быть от 95 до 200 п.н.; праймеры должны быть специфичны к максимальному количеству транскриптов гена.
Затем в амплификаторе BioRad CFX96 проводили ПЦР образцов с использованием интеркалирующего красителя SYBR® Green Master Mix (Bio-Rad Laboratories, Inc.; США) и олигонуклеотидных праймеров («Евроген»; Россия) (таблица). Уровень экспрессии генов интереса (GOI) оценивали относительно генов домашнего хозяйства (HKG) Gapdh и Actb. Расчет экспрессии в конкретной точке производился по формуле: Экспрессия гена = 2^[(Ct(HKG)-Ct(GOI)].
Иммуноблотинг
Образцы сетчатки пуллировали от двух животных, принадлежащих к одной группе.
После разделения в геле белки переносили методом полусухого электроблотинга на поливинилденфторидную мембрану Hybond-P (Cytiva; Великобритания), предварительно обработанную 100%-м метанолом, промытую водой MilliQ и замоченную в буфере для переноса, содержащем 25 мМ Трис, 0,15 мM глицина, 20% метанола. Помещали гель с плотно прижатой мембраной между двумя листами бумаги Wathman 3 ММ, смоченной в буфере для переноса, в аппарат для полусухого блотинга (GE Healthcare Amersham; США) и переносили белки на мембрану в течение 30 мин при токе 50 мА (1,2 мА на 1 см2). После электроблотинга промывали мембрану в
Трис-Твин-буфере (ТТБ; 50 мM Трис-HCl pH 7,4, 150 мМ NaCl, 0,1% Твин-20) 3 раза по 5 мин. Блокировали мембрану в 4%-м растворе обезжиренного сухого молока в ТТБ 1 ч при комнатной температуре, затем инкубировали с первичными антителами в том же растворе при 4 °C в течение ночи. После инкубации с первичными антителами промывали мембрану в ТТБ 3 раза по 5 мин и инкубировали с вторичными антителами 1,5 ч при комнатной температуре. После инкубации мембрану отмывали в ТТБ 3 раза по 5 мин.
Детекцию специфичного связывания антител проводили с помощью реагентов ECL Plus (Cytiva; Великобритания) согласно инструкции производителя. Для детекции хемилюминесции использовали рентгеновскую пленку. Количественный анализ результатов иммуноблотинга проводили с помощью денситометрического анализа, c использованием прибора BioSpectrum AC Chemi HR410 и программного обеспечения Vision Works LS (UVP; Великобритания). При проведении денситометрического анализа специфический сигнал от анализируемого белка нормализовали по отношению к сигналу от β-актина (после реинкубации мембраны с соответствующими антителами) для каждой дорожки отдельно.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
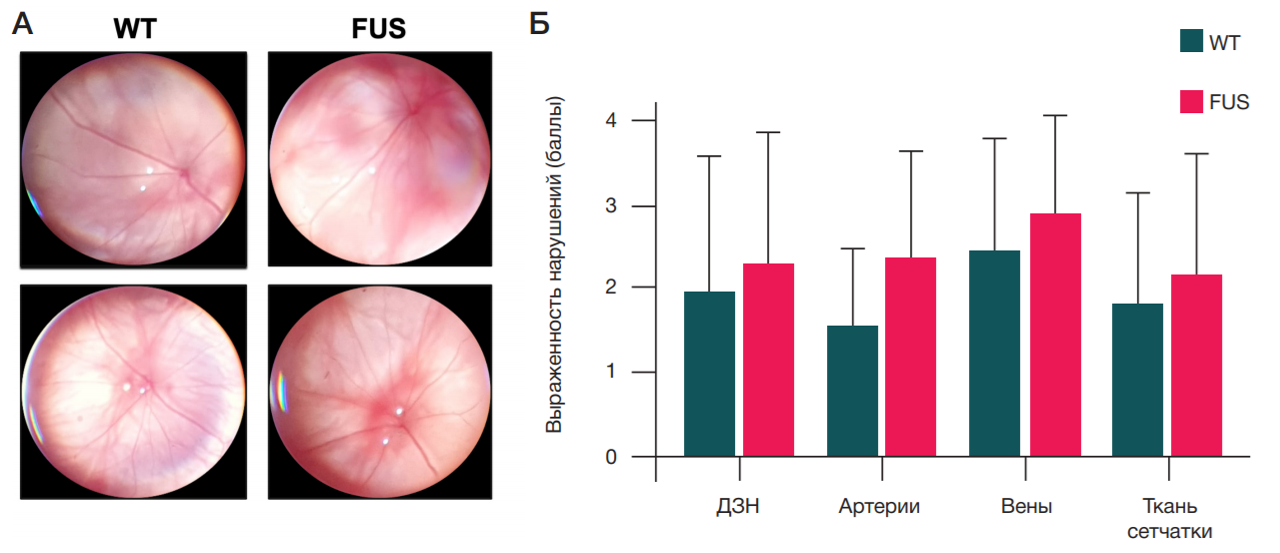
Трансгенные мыши FUS[1-359] демонстрируют умеренные офтальмоскопические аномалии
В обеих группах у части животных были обнаружены сосудистые аномалии и отечность диска зрительного нерва, что в целом характерно для мышей линии CD-1 [9, 10]. При статистическом анализе не выявлено достоверных различий между мутантными и дикотипными животными, однако у мышей FUS[1-359] отмечена тенденция к более выраженным нарушениям по всем исследуемым параметрам (рис. 1).
Аберрантный FUS не экспрессируется в сетчатке
Вестерн-блот-анализ не выявил наличие FUSиммунопозитивного сигнала в тканях сетчатки трансгенных мышей (рис. 2). Кроме того, при количественной ПЦР также не обнаружена экспрессия FUS на уровне мРНК. Таким образом, анализ экспрессии не подтвердил ретинальную экспрессию трансгена ни на транскриптомном, ни на белковом уровнях.
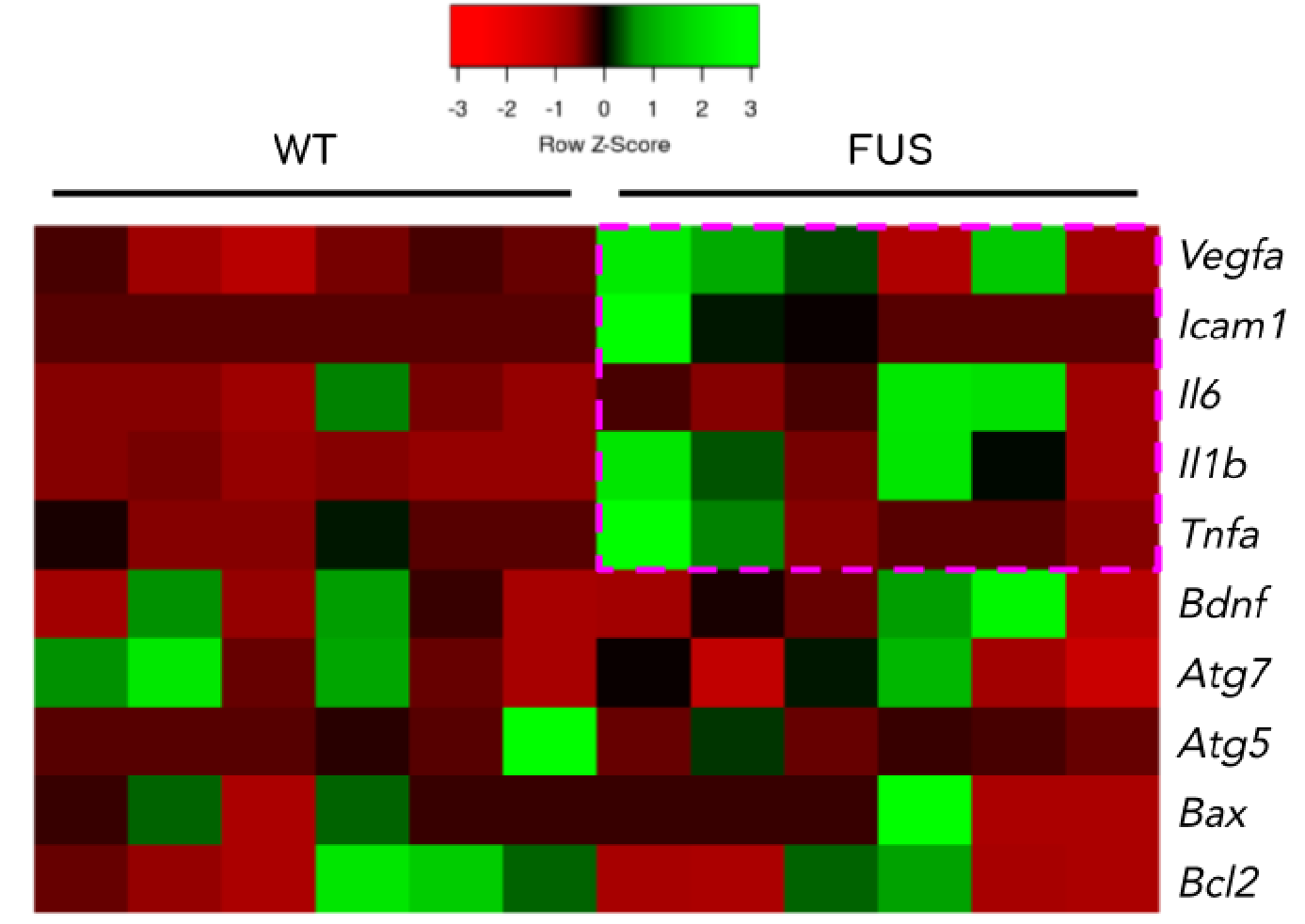
Трансгенные мыши FUS[1-359] характеризуются увеличенной экспрессией провоспалительных генов в сетчатке
При анализе ретинальной экспрессии таргетных генов было обнаружено, что в сетчатке трансгенных мышей FUS[1-359] происходит активация провоспалительных факторов Vegfa, Icam1, Il6, Il1b и Tnfa. Выраженных изменений экспрессии генов нейрорегенерации (Bdnf), аутофагии (Atg7, Atg5) и регуляции апоптоза (Bax, Bcl2) обнаружено не было (рис. 3).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Вовлечение сетчатки в патологический процесс было обнаружено при большинстве дегенеративных заболеваний центральной нервной системы, включая болезнь Альцгеймера [11], болезнь Паркинсона [12, 13] и лобно-височную деменцию [14]. Закономерно, что офтальмологические аномалии слабой степени выраженности также являются частым немоторным симптомом БАС [15]. Среди типичных клинических находок описаны нарушение цветового зрения [16], а также истончение сетчатки [17] и желтого пятна [18–20].
В нашем исследовании мы не обнаружили статистически значимых различий в выраженности патологических изменений сетчатки у исследуемых животных. Тем не менее по всем исследуемым параметрам была выявлена однозначная тенденция к более выраженным нарушениям у трансгенных мышей FUS[1-359]. Особенно яркие различия были обнаружены в отношении артериальной сети, что согласуется с результатами других исследователей [21].
Поскольку иммуноблотинг и количественная ПЦР показали, что трансген не экспрессируется в сетчатке, мы решили выяснить механизмы дегенерации сетчатки с помощью изучения активности наиболее общих молекулярных путей нейродегенеративного повреждения мотонейронов. В связи с этим в качестве основных таргетных мишеней мы выбрали гены, регулирующие воспаление, апотоз и аутофагию. Несмотря на то что вовлеченность путей аутофагии [22–24] и апоптоза [25] при БАС была подтверждена в исследованиях in vivo и in vitro, мы не нашли значимого отклонения в экспрессии генов Atg7, Atg5, Bax, Bcl2. Однако среди выбранных мишеней, была выраженно изменена экспрессия генов Vegfa, Icam1, Il6, Il1b и Tnfa, что указывает на роль воспаления в механизмах ретинальной дегенерации у мышей FUS[1-359]. Эти данные согласуются с предыдущими результатами, касающимися драматической роли воспалительной активации (и ее фармакологического подавления) в мышиной модели
FUS[1-359] [26, 27].
Таким образом, несмотря на отсутствие ретинальной экспрессии аберрантной формы белка FUS, трансгенные животные имеют признаки дегенеративных изменений сетчатки. Среди потенциальных механизмов повреждения заднего отрезка глаза можно выделить активацию микроглии [28], сосудистую регрессию [20], нейрофтальмологические взаимодействия через глимфатическую систему [29].
ВЫВОДЫ
Проведенное исследование продемонстрировало, что трансгенные мыши FUS[1-359] склонны к развитию структурных и функциональных аномалий глазного дна, несмотря на отсутствие экспрессии трансгена в сетчатке. Дальнейшие исследования природы выявленных нарушений могут актуализировать наиболее значимые механизмы FUS-ассоциированной ретинопатии.

- Yap TE, Balendra SI, Almonte MT, Cordeiro MF. Retinal correlates of neurological disorders. Ther Adv Chronic Dis. 2019; 10: 2040622319882205. doi:10.1177/2040622319882205
- Cerveró A, Casado A, Riancho J. Retinal changes in amyotrophic lateral sclerosis: looking at the disease through a new window. J Neurol. 2021; 268 (6): 2083–9. DOI: 10.1007/s00415-019-09654-w.
- Rojas P, de Hoz R, Ramírez AI, et al. Changes in Retinal OCT and their correlations with neurological disability in early ALS Patients, a Follow-Up Study. Brain Sci. 2019; 9 (12): 337. DOI: 10.3390/ brainsci9120337.
- Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015; 16 (6): 171. DOI: 10.4103/2152-7806.169561.
- Batra G, Jain M, Singh RS, Sharma AR, Singh A, Prakash A, et al. Novel therapeutic targets for amyotrophic lateral sclerosis. Indian J Pharmacol. 2019; 51 (6): 418–25. DOI: 10.4103/ijp.IJP_823_19.
- Zou ZY, Che CH, Feng SY, Fang XY, Huang HP, Liu CY. Novel FUS mutation Y526F causing rapidly progressive familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2021; 22 (1–2): 73–79. DOI: 10.1080/21678421.2020.1797815.
- Shelkovnikova TA, Peters OM, Deykin AV, et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 2013; 288 (35): 25266–274. DOI: 10.1074/jbc.M113.492017.
- Funikov SY, Rezvykh AP, Mazin PV, Morozov AV, Maltsev AV, Chicheva MM, et al. FUS(1-359) transgenic mice as a model of ALS: pathophysiological and molecular aspects of the proteinopathy. Neurogenetics. 2018; 19 (3): 189–204. DOI: 10.1007/s10048-018-0553-9.
- Mukaratirwa S, Petterino C, Naylor SW, Bradley A. Incidences and Range of Spontaneous Lesions in the Eye of Crl:CD-1(ICR) BR Mice Used in Toxicity Studies. Toxicol Pathol. 2015; 43 (4): 530–5. DOI: 10.1177/0192623314548767.
- De Groef L, Dekeyster E, Geeraerts E, Lefevere E, Stalmans I, Salinas-Navarro M, et al. Differential visual system organization and susceptibility to experimental models of optic neuropathies in three commonly used mouse strains. Exp Eye Res. 2016; 145: 235–47. DOI: 10.1016/j.exer.2016.01.006.
- Hart NJ, Koronyo Y, Black KL, Koronyo-Hamaoui M. Ocular indicators of Alzheimer's: exploring disease in the retina. Acta Neuropathol. 2016; 132 (6): 767–87. DOI: 10.1007/s00401-0161613-6.
- Mohana Devi S, Mahalaxmi I, Aswathy NP, Dhivya V, Balachandar V. Does retina play a role in Parkinson's Disease? Acta Neurol Belg. 2020; 120 (2): 257–65. DOI: 10.1007/s13760-020-01274-w.
- Huang L, Zhang D, Ji J, Wang Y, Zhang R. Central retina changes in Parkinson's disease: a systematic review and meta-analysis. J Neurol. 2020; 10. DOI: 10.1007/s00415-020-10304-9.
- Harrison IF, Whitaker R, Bertelli PM, O'Callaghan JM, Csincsik L, Bocchetta M, et al. Optic nerve thinning and neurosensory retinal degeneration in the rTg4510 mouse model of frontotemporal dementia. Acta Neuropathol Commun. 2019; 7 (1): 4. DOI: 10.1186/s40478-018-0654-6.
- Soldatov VO, Kukharsky MS, Belykh AE, Sobolev AM, Deykin AV. Retinal Damage in Amyotrophic Lateral Sclerosis:Underlying Mechanisms. Eye Brain. 2021; 13: 131–46. DOI: 10.2147/ EB.S299423.
- Boven L, Jiang QL, Moss HE. Diffuse colour discrimination as marker of afferent visual system dysfunction in amyotrophic lateral sclerosis. Neuroophthalmology. 2017; 41 (6): 310–4. DOI: 10.1080/01658107.2017.1326153.
- Rohani M, Meysamie A, Zamani B, Sowlat MM, Akhoundi FH. Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: a window to disease progression. J Neurol. 2018; 265 (7): 1557– 62. DOI: 10.1007/s00415-018-8863-2.
- Fawzi AA, Simonett JM, Purta P, et al. Clinicopathologic report of ocular involvement in ALS patients with C9orf72 mutation. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15 (7–8): 569–80. DOI: 10.3109/21678421.2014.951941.
- Ringelstein M, Albrecht P, Sudmeyer M, et al. Subtle retinal pathology in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2014; 1 (4): 290–7. DOI: 10.1002/acn3.46.
- Rojas P, Ramírez AI, Fernández-Albarral JA, López-Cuenca I, Salobrar-García E, Cadena M, et al. Amyotrophic Lateral Sclerosis: A Neurodegenerative Motor Neuron Disease With Ocular Involvement. Front Neurosci. 2020; 14: 566858. DOI: 10.3389/fnins.2020.566858.
- Crivello M, Hogg MC, Jirström E, Halang L, Woods I, Rayner M, et al. Vascular regression precedes motor neuron loss in the FUS (1-359) ALS mouse model. Dis Model Mech. 2019; 12 (8): dmm040238. DOI: 10.1242/dmm.040238.
- Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, et al. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proceedings of the National Academy of Sciences of the United States of America. 2017; 114: E8294–яE8303.
- Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiol Dis. 2019; 122: 35–40. DOI: 10.1016/j. nbd.2018.07.005.
- Strohm L, Behrends C. Glia-specific autophagy dysfunction in ALS. Semin Cell Dev Biol. 2020; 99: 172–82. DOI: 10.1016/j. semcdb.2019.05.024.
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014; 112: 24–49. DOI: 10.1016/j.pneurobio.2013.10.004.
- de Munter JPJM, Shafarevich I, Liundup A, Pavlov D, Wolters EC, Gorlova A, et al. Neuro-Cells therapy improves motor outcomes and suppresses inflammation during experimental syndrome of amyotrophic lateral sclerosis in mice. CNS Neurosci Ther. 2020; 26 (5): 504–17. DOI: 10.1111/cns.13280.
- Ninkina N. Stem cell therapy and FUS[1-359]-transgenic mice: A recent study highlighting a promising ALS model and a promising therapy. CNS Neurosci Ther. 2020; 26 (5): 502–3. DOI: 10.1111/ cns.13302
- Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The Role of Microglia in Retinal Neurodegeneration: Alzheimer's Disease, Parkinson, and Glaucoma. Front Aging Neurosci. 2017; 9: 214. DOI: 10.3389/fnagi.2017.00214.
- Dolzhikov AA, Bobyntsev II, Belykh AE, Shevchenko OA, Pobeda AS, Dolzhikova IN, et al. Pathogenesis of neurodegenerative pathology and new concepts of transport and metabolic systems of the brain and eye. Kursk Scientific and Practical Bulletin "Man and His Health". 2020; (1): 43–57. DOI: 10.21626/ vestnik/2020-1/06.