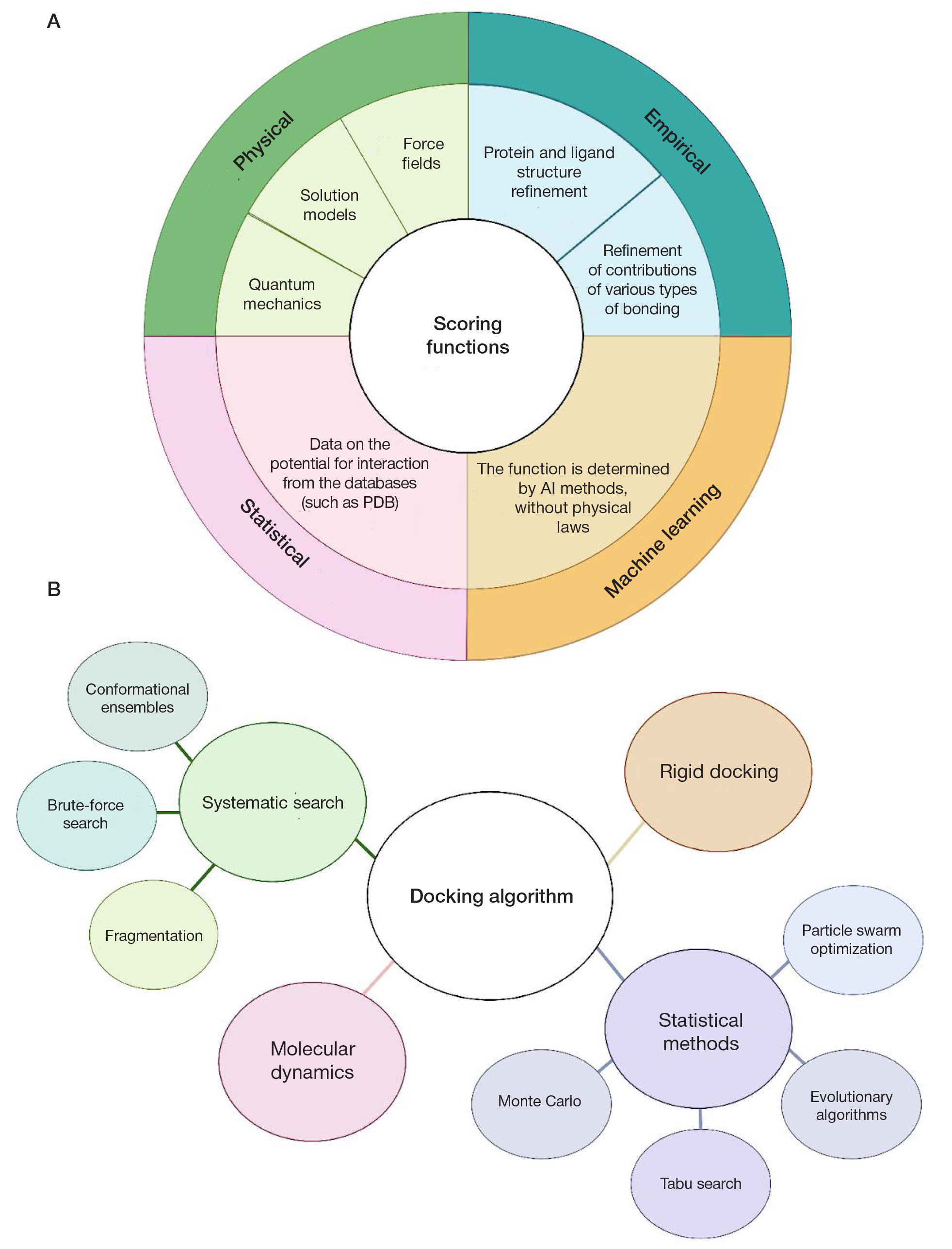
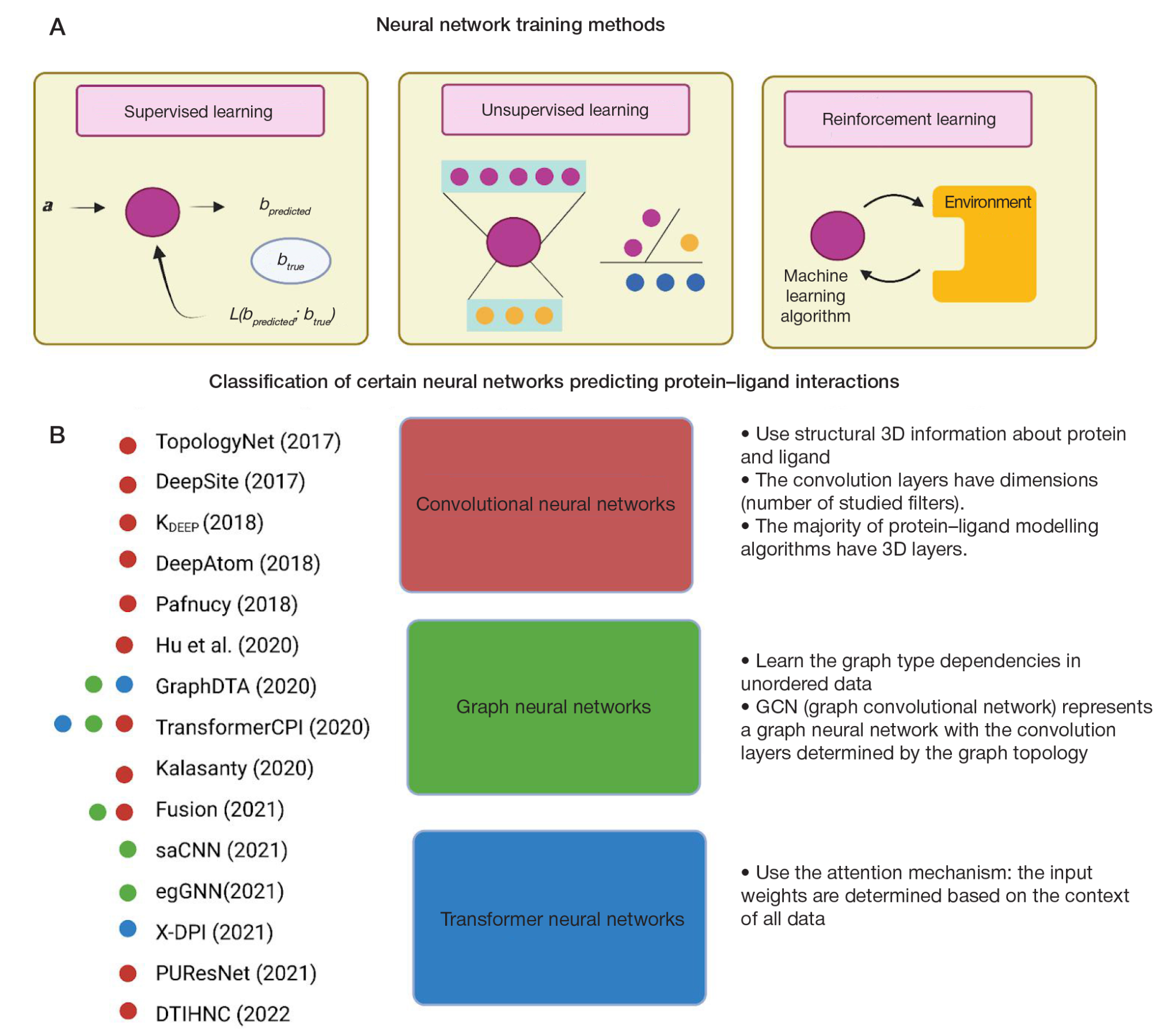
ISSN Print 2500–1094
ISSN Online 2542–1204
BIOMEDICAL JOURNAL OF PIROGOV UNIVERSITY (MOSCOW, RUSSIA)

1 Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia
2 Moscow Institute of Physics and Technology (MIPT), Dolgoprudny, Russia
3 Pirogov Russian National Research Medical University, Moscow, Russia
Correspondence should be addressed: Zinaida Mikhailovna Osipova
Miklukho-Maklaya, 16/10, Moscow, 117997, Russia; ur.hcbi@avoksakz
Funding: the study was supported by the Russian Science Foundation grant, project № 22-44-02024 (https://rscf.ru/project/22-44-02024/).
Author contribution: Barykin AD — literature review, manuscript writing, Chepurnykh TV — concept, literature review, manuscript writing and editing, Osipova ZM — project management, manuscript editing.